Patrice Koehl
Department of Computer Science
Genome Center
Room 4319, Genome Center, GBSF
451 East Health Sciences Drive
University of California
Davis, CA 95616
Phone: (530) 754 5121
koehl@cs.ucdavis.edu
|
|
| Patrice Koehl |
New Algorithms for Biological Data Modeling and Improved Simulations of Biological Processes |
|
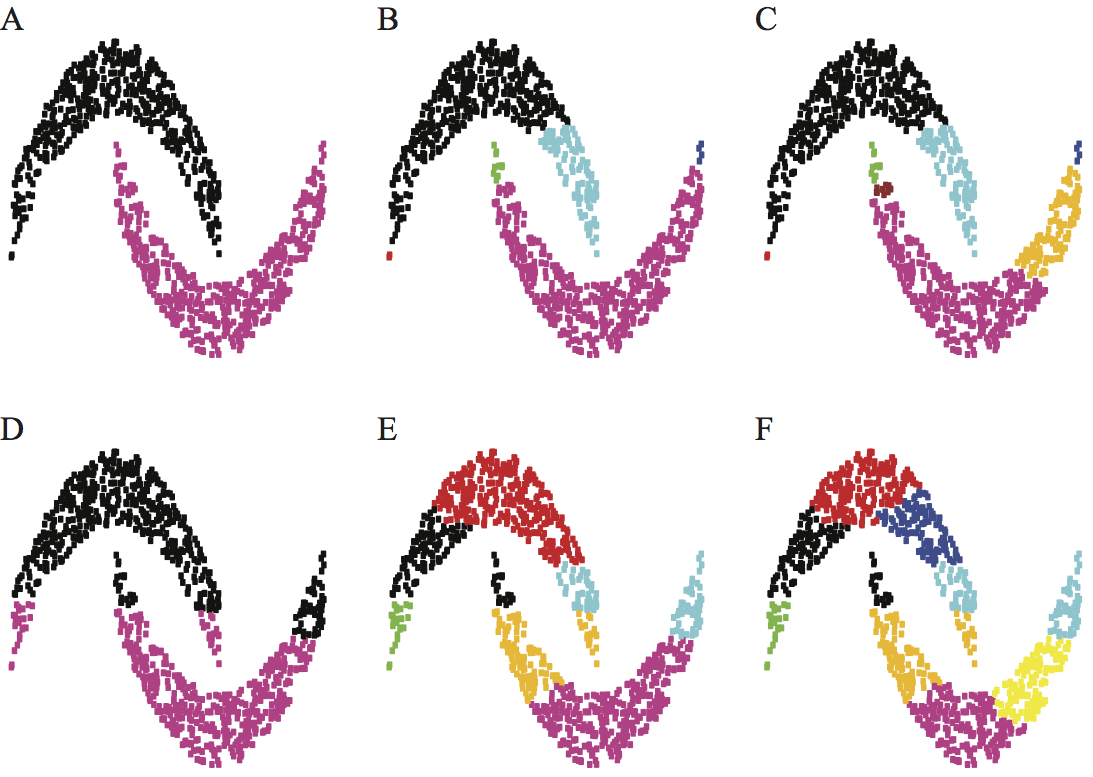
The ongoing transformation of biology to a quantitative discipline raises as many opportunities as challenges. The many -omics projects (genomics, proteomics, transcriptomics, metabolomics, to only name a few) allow us to map and identify all components of a living cell at the molecular level, from both a physical and functional standpoint. New technologies such as high resolution time-lapse microscopy and micro-scale devices have vastly enhanced our abilities to study the mechanics of biomolecules, cells, and tissues, giving us hope that we will be able to unravel the fundamentals of life. In addition to these technological advances that allow us to observe with better precision and improved scales, both in time and space, computational methods are playing an ever growing role in biology. As physical models improve and greater computational power becomes available, simulation of complex biological processes will become increasingly tractable. The challenges however come in analyzing and interpreting the vast amount of data generated from these disciplines. These data are complex in size, dimension, and heterogeneity. As a recent (Feb 12, 2013) MIT Technology Review reports, the challenge "is not processing or storing this amount of data- Moore's law should take care of all that. Instead, the difficulty is uniquely human. How do humans access and make sense of the exascale data sets?" We must develop new methods for extracting knowledge from data, as well as new simulation methods that allow us to implement this knowledge into holistic models that will enable understanding. I am playing my own part in this research effort by developing new mathematical and computational methods for extracting knowledge from data as well as for pushing the limits of physical simulation methods. I provide two examples below, one related to clustering, and the other to solving complex partial differential equations. Clustering: understanding the underlying geometry of data setsMain collaborator: Fushing Hsieh (Statistics, UC Davis) The advent of high-throughput technologies and the concurrent advances in information sciences have led to a explosion in size and complexity of the data sets collected in biological sciences. The biggest challenge today is to assimilate this wealth of information into a conceptual framework that will help us decipher biological functions. A large and complex collection of data, usually called a data cloud, naturally embeds multi-scale characteristics and features, generically termed geometry. Understanding this geometry is the foundation for extracting knowledge from data. We have developed a new methodology, called data cloud geometry (DCG) to resolve this challenge. Briefly, this algorithm proceeds as follows. The empirical relational measure is transformed into a temperature-regulated potential defined on the links between the nodes. Based on this potential, we extract at very low temperature a collection of motifs, which become building blocks for growing clusters via data-driven merging dynamics as temperature is being raised slowly. A series of phase transitions on this merging dynamics is identified at a series of critical temperatures. These temperatures are then taken as energy barrier heights to define an ultrametric topology onto the data cloud as it is a system on a ground state. This topology provides measurable and natural distances between clusters. In comparison to our proposed approach here, the widely used Hierarchical Clustering (HC) technique requires that the underlying distance metric is robust with respect to errors in the measures, and that the "true" clusters are well separated and more or less in ball shape. Given that most empirical distance measures and data geometries do not fulfill these conditions simultaneously, our resolving protocol circumvent these problems by transforming any such measure into a robust ultrametric in a data-driven fashion, as well derives a natural distance measure between clusters of data. The figure below shows the effectiveness of our method compared to HC on an artificial data set.
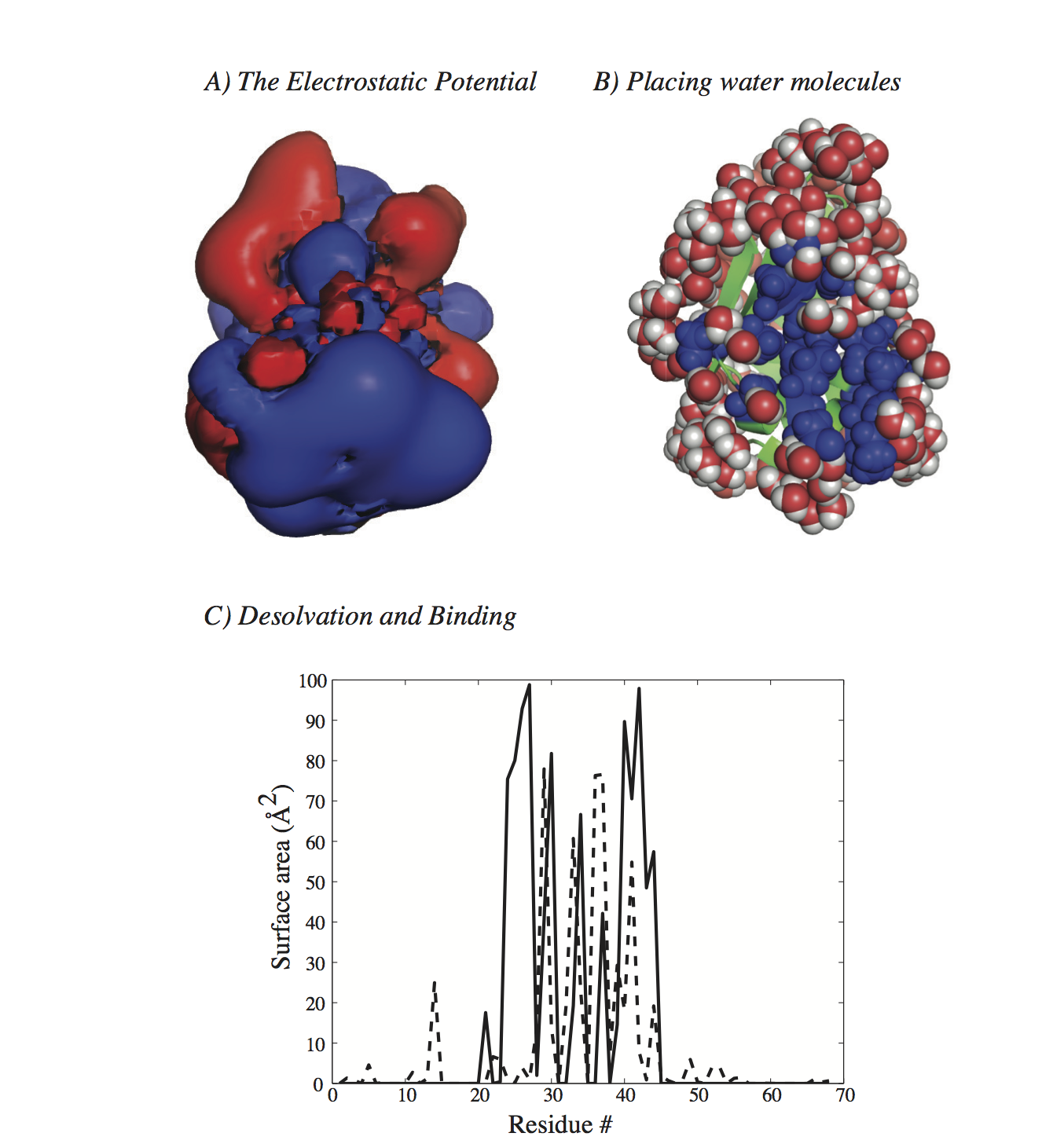
Related paper: H. Fushing, H. Wang, K. VanderWaal, B. McCowan, and P. Koehl. Multi-scale clustering by building a robust and self correcting ultrametric topology on data points. PLoS One, 8:e56259, 2013. Solving PDEs: the Dipolar Poisson-Boltzmann Langevin equationMain collaborators: Marc Delarue (Institut Pasteur, France), Henri Orland (CEA, Paris, France) The Poisson-Boltzmann formalism is among the most popular approaches to modeling the solvation of molecules. It assumes a continuum model for the water, leading to a dielectric permittivity that only depends on position in space. In contrast, the Dipolar Poisson-Boltzmann Langevin (DPBL) formalism represents the solvent as a collection of orientable dipoles with non uniform concentration; this leads to a non linear permittivity function that depends both on the position and on the local electric field at that position. The differences in the assumptions underlying these two models lead to significant differences in the equations they generate. The PB equation is a second order, elliptic, nonlinear partial differential equation (PDE). Its diffusion coefficients correspond to the dielectric permittivity and are therefore constant within each sub-domain of the system considered (i.e. inside and outside of the molecules considered). While the DPBL equation is also a second order, elliptic, nonlinear PDE, its response coefficients are non linear functions of the electrostatic potential. Many solvers have been developed for the PB equation; to our knowledge, none of these can be directly applied to the DPBL equation. The methods they use may adapt to the difference; their implementations however are PBE specific. We have adapted the PBE solver originally developed by Holst and Saied (J. Comput. Chem, 16, 337-364, 1995) to the problem of solving the DPBL equation. This solver uses a truncated Newton method with a multigrid pre-conditioner. Numerical evidences suggest that it converges for the DPBL equation, and that the convergence is superlinear. It is found however to be slow and greedy in memory requirement for problems commonly encountered in computational biology and computational chemistry. To circumvent these problems, we propose two variants, a quasi Newton solver based on a simplified, inexact Jacobian and an iterative self-consistent solver that is based directly on the PBE solver. While both methods are not guaranteed to converge, numerical evidences suggest that they do and that their convergence is also superlinear. Both variants are significantly faster than the solver based on the exact Jacobian, with a much smaller memory footprint. All three methods have been implemented in a new code named AquaSol, which is freely available (send me an e-mail). The figure below shows one application of DPBL to study the solvation of a protein.
Related paper: P. Koehl and M. Delarue. AQUASOL: an efficient solver for the dipolar Poisson-Boltzmann-Langevin equation. J. Chem. Phys., 132:064101, 2010.
|