Patrice Koehl
Department of Computer Science
Genome Center
Room 4319, Genome Center, GBSF
451 East Health Sciences Drive
University of California
Davis, CA 95616
Phone: (530) 754 5121
koehl@cs.ucdavis.edu
|
|
| Patrice Koehl |
Geometry and Topology of Biological Systems |
Finding efficient algorithms to describe, measure and compare shapes is a central problem in image processing. This problem arises in numerous disciplines that generate extensive quantitative and visual information. Among these, biology occupies a central place. For example, registration of brain anatomy is essential to many studies in neurobiology. In parallel, the belief in molecular biology that the structure (or shape) of a protein is a major determinant of its function has led to the development of many methods for representing, measuring and comparing the shapes of protein structures. In the following, I show two examples of my recent research efforts in these two directions.
Main collaborators: Joel Hass (UC Davis), Nina Amenta (UC Davis), Owen Carmichael (UC Davis)
|
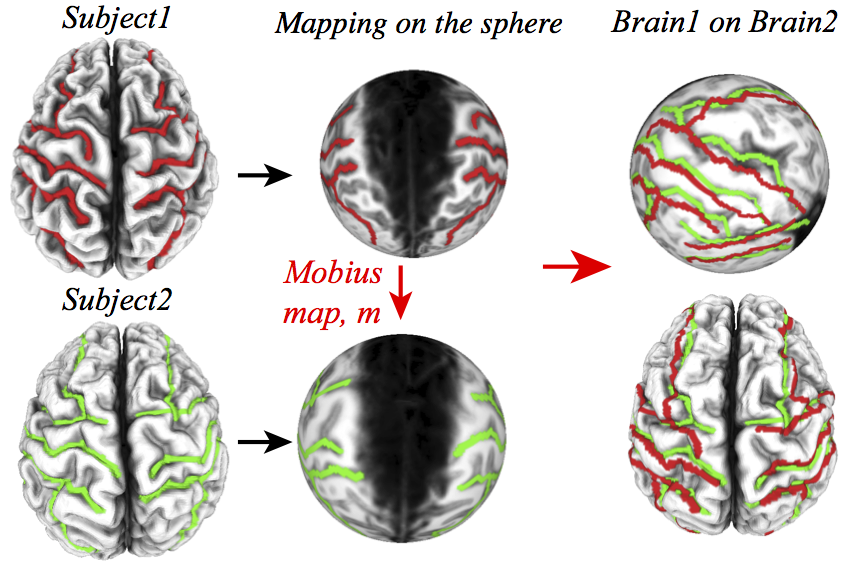
In general, methods that compare shapes can be classified into two categories: those that derive features (also called shape descriptors) for each shape separately that can then be compared using standard distance functions, and those that directly attempt to map one shape onto the other, thereby providing both local and non-local elements for comparison. I am currently interested in developing methods that generate mappings between two shapes that are defined by surfaces of genus zero. In collaboration with Prof Joel Hass, Mathematics Department, UC Davis, we have recently proposed a new algorithm for shape registration based on the idea of a globally optimal conformal mapping between two surfaces of genus zero. In this approach, the whole mesh representing the source surface is warped onto the target surface, using the mapping defined through the composition of discrete conformal mappings of the surfaces onto the sphere and the Möbius transformation between these mappings. The Möbius transformation is then optimized to lead to minimal distortion between the source mesh and its image, where distortion is measured as difference from isometry. The optimization chooses a Möbius transformation that minimizes a measure of distortion by gradient descent from a carefully chosen initial value. The figure below illustrates this procedure for the problem of aligning the brains of two human subjects. |
|
|
Automatic brain surface mappingThe surfaces of two brains from human subjects are shown on the left. The sulcal structures are color-coded. The best registration (using the method described above) is shown on the right, both on the surface of the sphere (side view) and on the surface of brain 2 (top view). The colored sulci are shown to illustrate the quality of this registration. |
Related papers:
Main collaborator: Herbert Edelsbrunner (IST Austria)
Significance of the shapes of biomoleculesThe idea that shape defines function is a general concept from physical chemistry. Molecular structure or shape and chemical reactivity are highly correlated as the latter depends on the positions of the nuclei and electrons within the molecule. Indeed, chemists have long used three-dimensional plastic and metal models to understand the many subtle effects of structure on reactivity and have invested in experimentally determining the structures of important molecules. A common concrete model representing molecular shape is a union of balls, in which each ball corresponds to an atom. Properties of the molecule are then expressed in terms of properties of the union. For example, the putative active sites of an enzyme are detected as cavities and the interaction between a protein and its environment is quantified through the surface area and/or volume of the union of balls. The most common use of molecular shape however is found in the quantification of the hydrophobic effect. For this, Lee and Richards introduced in 1971 the concept of the solvent-accessible surface. They computed the accessible area of each atom in both the folded and extended state of a protein, and found that the decrease in accessible area between the two states is greater for hydrophobic than for hydrophilic atoms. These ideas were refined by Eisenberg and McLachlan, who introduced the concept of a solvation free energy for large biomolecules, computed as a weighted sum of the accessible areas of all their atoms i. It is not clear, however, which surface area should be used to compute this solvation energy. There is also some evidence that for small solutes, the hydrophobic effect is not proportional to the surface area, but rather to the solvent excluded volume of the molecule. Current models for the non-polar part of the solvent energy include both a surface-based term and a volume-based term. Within this debate on the exact form of the solvation energy, there is however a consensus that it depends on the geometry of the biomolecule under study, more specifically on its volume and surface area.
|
|
|
|
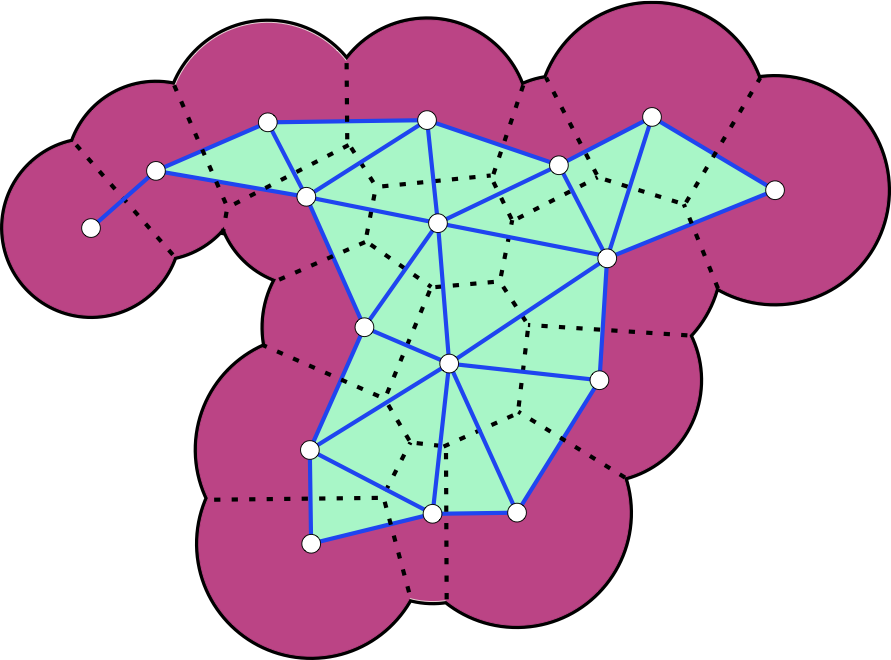
Voronoi decomposition and dual complexGiven a finite set of disks, the Voronoi diagram decomposes the plane into regions, one per disk, such that any point in the region assigned to disk Si is closer to that disk than to any other disk. In the drawing, we restrict the Voronoi diagram (dashed lines) to within the portion of the plane covered by the disks (magenta) and get a decomposition of the union into convex regions. The dual Delaunay triangulation is obtained by drawing edges between circle centers of neighboring Voronoi regions. The dual complex is a subset of the Delaunay triangulation, limited to the edges and triangles (light blue) whose corresponding Voronoi regions intersect within the union of disks. |
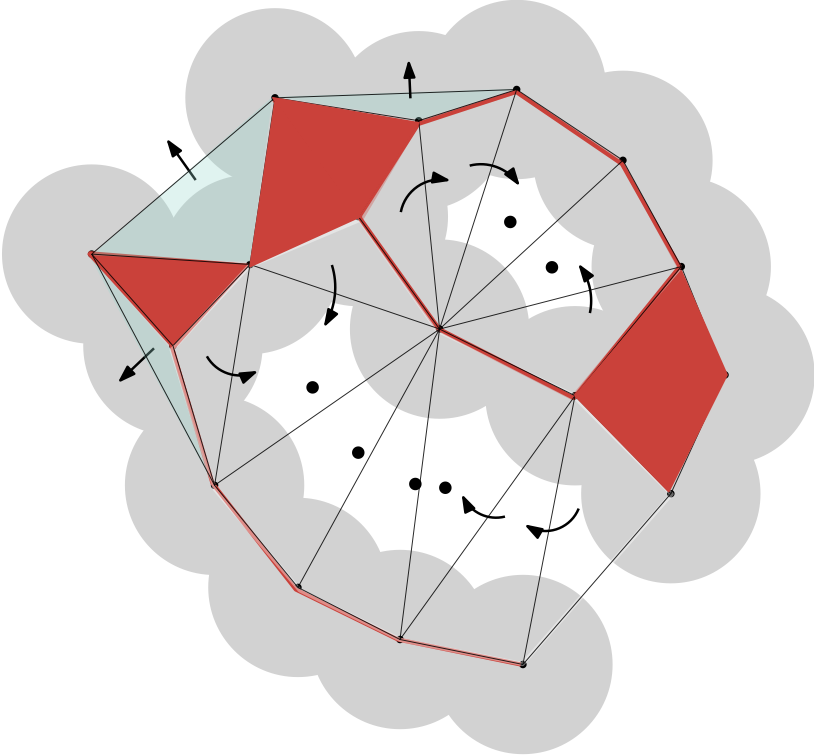
Discrete flow and pockets in a union of disksThe dual complex of the union of disks is shown in red; all triangles in the Delaunay complex that do not belong to the dual complex are referred to as empty. Acute empty triangles contain their orthocenters: they correspond to sinks. We identify them with large dots to mark the position of the orthocenter. The obtuse empty triangles either flow to these acute triangles or to the outside ("infinity"). Triangles III, IV and V for example flow to infinity: they do not define pockets. The remaining triangles can be partitioned into two groups: region I is completely surrounded by the union of disks and therefore defines a void, while region II is connected to the outside by one mouth, and is referred to as a pocket. |
Pavani and Ranghino proposed in 1982 an analytical method for computing the volume of a molecule based on the inclusion-exclusion formula. In their implementation, only intersections of up to three balls were considered. Petitjean however noticed that practical situations for proteins frequently involve simultaneous overlaps of up to six balls. Subsequently, Pavani and Ranghino's idea was generalized to any number of simultaneous overlaps by Gibson and Scheraga in 1987 and by Petitjean, applying a theorem by Kratky that states that higher-order overlaps can always be reduced to lower-order overlaps. Doing the reduction correctly remains however computationally difficult and expensive. The Alpha Shape Theory proposed by Herbert Edelsbrunner solves this problem using Delaunay triangulations and their filtrations. I have been collaborating with Prof. Edelsbrunner (IST, Austria) to extend his method to compute derivatives of the measures of a molecule, as well as to implement the Alpha Shape theory such that it can be applied to systems of arbitrary size, with up to millions of balls.
|
|
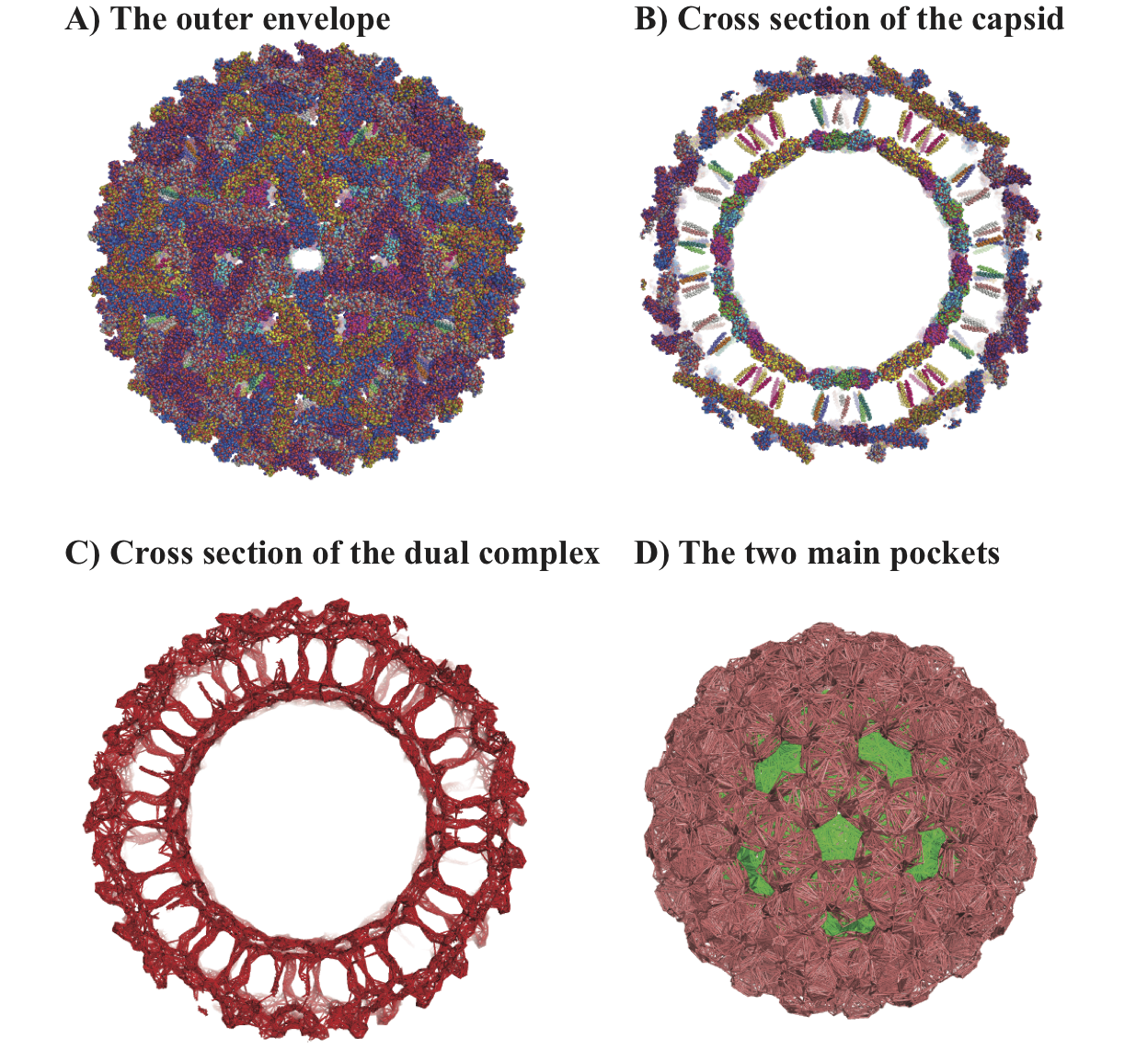
Characterizing the geometry of the Sindbis virusThe Sindbis virus is an RNA virus, member of the alphavirus; it is transmitted by mosquitoes and is responsible for the Sindbis fever, most common is South and East Africa, the Philipines and Australia. The structure of the full virion consists of two protein capsids (the outside capsid made of glycoproteins and the inner nucleocapsid, a lipid bilayer sandwiched between the two capsids, a set of transmembrane domains that cross the lipid bilayer and connect the two capsids, and the single stranded RNA that occupy the cavity inside the nucleocapsid; it was determined by combination of X-ray crystallography on individual proteins of the capsid and cryoelectron microscopy. All images show here are based on the complete structure of the capsids obtained from the VIPERdb2 database (file 1ld4.vdb); note that this file only includes the proteins (capsids and transmembrane domains). A. Surface of the virus: the outer glycoprotein capsid. B. Cross section through the capsid, showing the outer capsid, the inner nucleocapsid and the transmembrane. C. Cross section of the dual complex corresponding to the two capsids and transmembrane domains. The simplices of this complex define all the terms of the inclusion-exclusion formula needed to compute the volume and surface area of the virus. D. The two main pockets identified by UnionBall: the central pocket (in green) occupies the whole region in the center; it includes many large tetrahedra that are cut by the cross section. The second largest pocket (shown in pink) between the two capsids corresponds to the region where the lipid bilayer is found. |
Related papers:
| Page last modified 18 July 2017 | http://www.cs.ucdavis.edu/~koehl/ |